Zaburzenia depresyjne stanowią jeden z „najpopularniejszych” zaburzeń zdrowotnych na świecie – dotyka ok. 10-15% populacji rocznie i są trzecią najczęstszą przyczyną prowadzącą do niesprawności i/lub niezdolności do pracy. Spośród chorób mózgu ponad 60% kosztów społecznych i ekonomicznych generują zaburzenia psychiczne, głównie zaburzenia depresyjne i lękowe. Kobiety dwa razy częściej niż mężczyźni zapadają na zaburzenia depresyjne. Globalne statystyki i wszelkie wykresy sprzed ostatnich 29 lat są dostępne między innymi na bazie danych Institute for Health Metrics and Evaluation (IHME) – GBD Compare GLOBAL Data Visualization IHME.

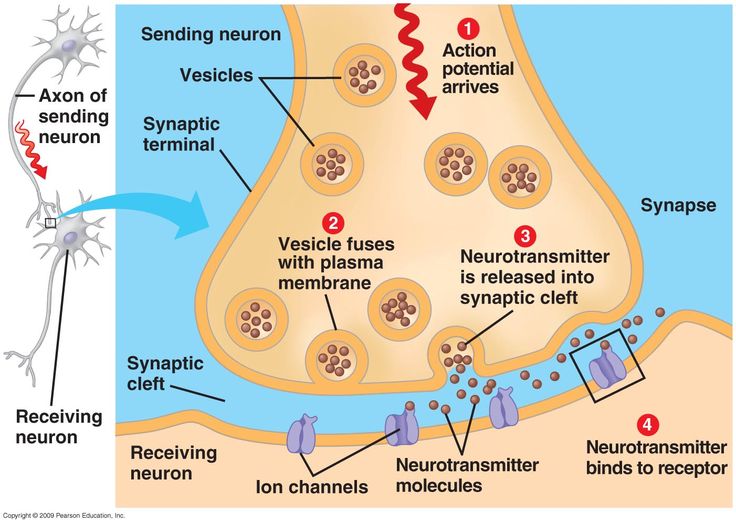
W ciągu wielu lat zrozumienie depresji i jej diagnoza koncentrowały się głównie wokół hipotezy monoaminowej, a jej leczenie i większość leków przeciwdepresyjnych ma na celu zwiększenie ilości neuroprzekaźników: serotoniny i/lub norepinefryny w szczelinie synaptycznej. Pytanie jednak czy niska serotonina i/lub noradrenalina lub inne neuroprzekaźniki jak dopamina czy GABA rzeczywiście powodują depresję pozostaje dalej bez odpowiedzi, gdyż liczne próby naukowców w znalezieniu tej odpowiedzi zawiodły, a leczenie do tej pory jest subiektywne, gdyż opiera się na objawach, a nie na zmierzonych zaburzeniach równowagi biochemicznej mózgu (ze względu na brak narzędzi do zrobienia tego).
Powszechnie wiadomo, że ok. 30% pacjentów nie reaguje poprawą na leki antydepresyjne, a wśród 70% osób wykazujących znaczną poprawę samopoczucia (mierzoną Skala Depresji Hamiltona), wymagają dodatkowych interwencji psychospołecznych w celu osiągnięcia całkowitej remisji – czyli według doniesień naukowych tylko ok 1/3 osób doświadcza remisji za pomocą leków, a ok. 50-66% osób z zaburzeniami depresyjnymi wymagają dodatkowych interwencji.
I choć zaburzenia czy to w poziomach, czy wychwycie zwrotnym czy sygnalizacji receptorowej neurotransmiterów w zaburzeniach depresyjnych jest kwestią niepodważalną, to jednak na dzień dzisiejszy wiemy na pewno, że nie możemy skupiać całą uwagę tylko na powyższych aspektach, gdyż zaburzenia depresyjne są wieloczynnikowym stanem chorobowym w etiologii którego występuję ścisła interakcja pomiędzy tzw. genami podatności oraz czynnikami środowiskowymi. Warto też pamiętać o tym, że depresja może być zarówno pierwotna jak i wtórna (po innym stanie chorobowym czy po stosowanych środkach farmakologicznych) oraz o tym, że diagnozę może postawić tylko i wyłącznie lekarz!!!
Próby identyfikacji konkretnych genów lub mutacji genetycznych odpowiedzialnych za depresję spotkały się z licznymi ograniczeniami. Kilka obszernych badań (całego genomu, badania rodzinne i bliźniacze) wskazują na to, iż różne genetycznie predysponowane do depresji przypadki oraz rodzaje zaburzeń depresyjnych mogą być warunkowane:
– mutacjami występującymi jednocześnie w różnych genach;
– ich wzajemnych interakcji;
– oraz tego w jaki sposób oddziałuje na nas środowisko (czynnik epigenetyczny).
Szeroki podział zaburzeń depresyjnych oraz fakt, że istnieje wiele potencjalnych przyczyn mogących do nich doprowadzać jest tematem rzeką, dlatego chciałabym dziś się skupić tylko na jednym aspekcie – stanach zapalnych.
W badaniu ludzkiego genomu również zostały zidentyfikowane geny, które są związane z funkcjonowaniem układu odpornościowego oraz powodują powstanie tzw. fenotypu prozapalnego. Prozapalny fenotyp stwarza potencjalny genetyczny fundament zwiększonego ryzyka rozwoju zaburzeń depresyjnych.
Układ odpornościowy odgrywa ważną rolę w rozwoju i funkcjonowaniu mózgu. Aktywacja układu odpornościowego jest naturalną, ważną i potrzebną reakcją organizmu na uszkodzenie tkanek czy różnego rodzaju zakażenia drobnoustrojami chorobotwórczymi. Jednak, kiedy reakcja zapalna wychodzi spod kontroli, i z ostrego stanu zapalnego staje się przewlekłym stanem zapalnym to może doprowadzić do rozwoju wielu chorób przewlekłych, w tym układu nerwowego. Warto wspomnieć o tym, że w porównaniu do ostrego stanu zapalnego objawy przewlekłego stanu zapalnego są bardzo niespecyficzne i mało charakterystyczne.
Wraz z odkryciem zwiększonego poziomu cytokin prozapalnych u ludzi z depresją ponad dwie dekady temu, pojawia się coraz więcej dowodów naukowych na to, że zmiany w pracy układu immunologicznego biorą udział w patogenezie depresji.
Hipoteza depresji wywołana cytokinami prozapalnymi (tzw. teoria zapalna depresji) pojawiła się wraz z obserwacją, że:
1. Leczenie chorób terapią cytokinową/immunoterapia (interferonem alfa lub IL-2) może wywołać objawy depresji, a u ok. 40% osób się rozwija kliniczna depresja po wspomnianym leczeniu;
2. Choroby o podłożu zapalnym są związane ze zwiększonym odsetkiem zapadalności na wtórną depresję niż choroby niezapalne – depresja ogólnie występuje częściej u osób z chorobami, które się wiążą z dysfunkcją immunologiczną. Wyższy wyjściowy poziom stanów zapalnych wiąże się z brakiem odpowiedzi na farmakologiczne leczenie przeciwdepresyjne – czyli powstanie depresji lekoopornej. Cytując pracę „Teoria zapalna depresji – najważniejsze fakty” : „Choroby układu krążenia, choroby układu oddechowego, choroby metaboliczne i autoimmunologiczne są nie tylko czynnikami ryzyka wystąpienia epizodu depresyjnego, ale również uznawane są za czynnik lekooporności depresji i jedną z istotnych przyczyn nawrotowości epizodów obniżonego nastroju”;
3. Jedna trzecia osób z depresją wykazuje podwyższony poziom cytokin zapalnych bez obecności innej/równoległej choroby, która by te stany zapalne powodowała;
4. U wielu pacjentów z depresją obserwuje się nadmierną aktywację układu odpornościowego – zarówno depresja, jak i szereg chorób cywilizacyjnych (nadciśnienie, choroba wieńcowa, cukrzyca) mają wspólne tło immunologiczne;
5. Liczne badania naukowe potwierdzają zwiększoną ilość cytokin prozapalnych w depresji, takich jak interleukiny IL-1beta i IL-6, czynnik martwicy nowotworów alfa (TNF-alfa) oraz interferon gamma (IFN-gamma);
6. Kilka cytokin prozapalnych może nadmiernie aktywować układ noradrenergiczny oraz oś podwzgórze-przysadka-nadnercza (HPA), które właśnie często są nadmiernie pobudzone u pacjentów z depresją nawet bez obecności przewlekłego stanu zapalnego – zaburzenia w obrębie osi HPA dotyczą 50–75% pacjentów ze zdiagnozowaną dużą depresją ( z angl. Major depression), a zaburzenia depresyjne pod względem patofizjologicznym przypominają stres przewlekły.
Nasuwa się pytanie w takim razie co było pierwsze?
Czy najpierw były stany zapalne które przyśpieszyły zachorowanie na depresje czy depresja spowodowała stan zapalny i rozwój choroby z tłem zapalnym, która dodatkowo nasiliła przebieg depresji?
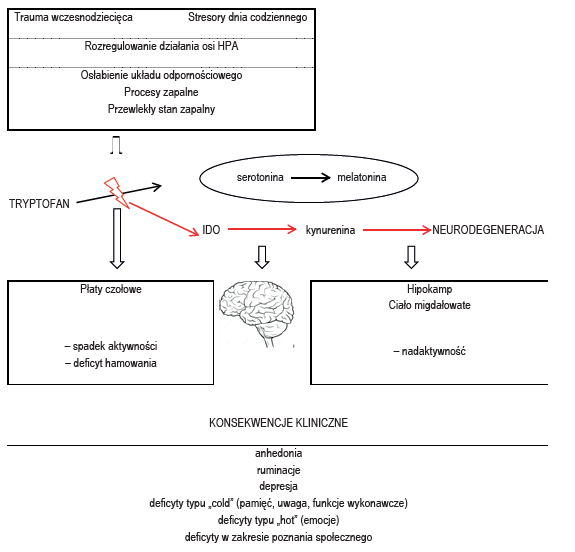
Ani jedno, ani drugie – depresja i stan zapalny wpływają na siebie dwukierunkowo i trzeba to rozpatrywać trochę inaczej. Dr. hab. med. prof. nadzw. Piotr Gałecki jest jednym z niewielu osób w Polsce, który zajmuje się współzależnością występowania depresji razem z innymi chorobami przewlekłymi z tłem zapalnym. Profesor uważa, że biorąc pod uwagę wspólne tło immunologiczne to dla obu jednostek chorobowych, czyli dla depresji oraz wszystkich chorób cywilizacyjnych o tym co rozwinie się wcześniej (depresja czy któraś z chorób cywilizacyjnych) decydują czynniki środowiskowe oraz psychologiczne (komponent osobowościowy). Cytując słowa profesora: „Choroby zapalne i depresja mają wspólny czynnik ryzyka – jest to mobilizacja układu immunologicznego. Infekcje, które pojawią się przy leczeniu depresji, powodują pogorszenie stanu psychicznego i rokowania dla depresji. Dzieje się tak, ponieważ cytokiny prozapalne zmniejszają produkcję serotoniny, przekierowując metabolizm tryptofanu oraz, co ważne, wpływają bezpośrednio na neurogenezę w układzie limbicznym”.
Chciałabym się odnieść do tego zdania: „cytokiny prozapalne zmniejszają produkcję serotoniny, przekierowując metabolizm tryptofanu…”. Chodzi o to, że wzrost poziomu cytokin zapalnych tak samo jak nadmierna aktywność osi podwzgórze-przysadka-nadnercza są źródłem nieprawidłowości w działaniu szlaku kynureninowego. Powyższe czynniki powodują zwiększoną aktywność enzymu obecnego w mikrogleju, astrocytach oraz neuronach – indoloamino-2,3-dioksygenazy (IDO), co zwiększa konwersję tryptofanu w kynureninę oraz kwas chinolinowy, czyli neurotoksyczne substraty zwiększające ryzyko wystąpienia procesów neurodegeneracyjnych oraz neurotoksycznych. W ten sposób IDO zmniejsza ilość tryptofanu dostępną do produkcji serotoniny, ale nie tyle niedobór tryptofanu, co substancje neurotoksyczne powstające w wyniku nadmiernej aktywacji tego szlaku mogą być źródłem depresji (patrz powyższe zdjęcie).
Teoria zapalna depresji rozpatruje ją jako zaburzenie psychoneuroimmunologiczne.
Warto na koniec oczywiście powiedzieć o tym, że nie wszyscy chorzy cierpią na depresję, tak samo jak nie wszyscy z depresją mają zwiększone poziomy cytokin prozapalnych. Niemniej jednak, regulacja pracy układu odpornościowego oraz adresowanie stanów zapalnych może być po prostu jednym z bardzo ważnych elementów całej układanki, szczególnie wśród osób, które nie reagują dobrze na leki antydepresyjne.
Co możemy zrobić by zmniejszyć ryzyko depresji o podłożu zapalnym?
1. Przede wszystkim udać się po profesjonalną pomóc, jeśli występują u Ciebie objawy depresji wedle: skali depresji Hamiltona, inwentarza depresji Becka (BDI), skali Montgomery-Asberg albo ICD-10;
2. W drugiej kolejności – jeśli występuję u Ciebie choroba o podłożu zapalnym (np. autoimmunizacyjna, insulinooporność itd) to „zająć się” w pierwszej kolejności tym;
3. SEN! o tym nigdy nie przestanę trąbić, ale bez wystarczającej ilości oraz jakości snu jak i dbania o jego higienę daleko nie zajedziemy;
4. Pracować nad sposobem w jaki radzimy sobie z codziennym stresem (polecam zapoznać się z triada poznawcza Aarona Becka oraz pracować nad tym za pomocą terapii poznawczo-behawioralnej) oraz zmniejszyć poziom obciążenia allostatycznego (tu mogę odesłać do genialnej książki Dlaczego Zebry Nie Mają Wrzodów);
5. Zadbać o oś mózgowo-jelitową wraz z mikroflora jelitowa, lepiej się odżywiać i unikać diety prozapalnej. Żywienie ma być oczywiście zawsze dostosowane indywidualnie, ale na początek dieta na wzór środziemnomorskiej może okazać się pomocna (przynajmniej na razie wypada bardzo dobrze w wielu badaniach naukowych);
6. Trenować regularnie, ale rekreacyjnie – już chyba wiemy niestety, że sport wyczynowy niema nic wspólnego ze zdrowiem i nadmiar treningów wraz z zaniedbywaniem aspektów regeneracyjnych może powodować prawdziwe piekło prozapalne.
7. Suplementacja? Hmm, może okazać się w ogóle zbędna kiedy zadbamy o powyższe punkty, a jeśli już to ma być celowana w konkretny przypadek, dlatego celowo umieszczam na ostatnim miejscu i nie zgłębiam się w szczegóły.
Więcej na ten temat się znajduje w grudniowym (2018) oraz styczniowym (2019) numerach czasopisma FOOD FORUM.
* Wpis ma charakter informacyjny. Wyniki badań i dolegliwości warto konsultować z lekarzem.
BIBLIOGRAFIA:
1. Jeffrey R. Lacasse and Jonathan Leo: Serotonin and Depression: A Disconnect between the Advertisements and the Scientific Literature. PLoS Med. 2005 Dec; 2(12): e392.
2. Berton O, Nestler E.J.: New approaches to antidepressant drug discovery: beyond monoamine. Nature Reviews Neuroscience 2006, volume 7, pages 137–151.
3. Khalid Saad Al-Harbi: Treatment-resistant depression: therapeutic trends, challenges, and future directions. Patient Prefer Adherence. 2012; 6: 369–388.
4. Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium: A mega-analysis of genome-wide association studies for major depressive disorder.Mol Psychiatry. 2013 Apr;18(4):497-511.
5. MSc Yanni Zeng, PhD Pau Navarro, et.al.: Shared Genetics and Couple-Associated Environment Are Major Contributors to the Risk of Both Clinical and Self-Declared Depression. EBioMedicine, December 2016, Volume 14, Pages 161–167.
6. Bernhard Luscher,Qiuying Shen: The GABAergic Deficit Hypothesis of Major Depressive Disorder.Mol Psychiatry. 2011 Apr; 16(4): 383–406.
7. López León S, Croes EA, et.al.: The dopamine D4 receptor gene 48-base-pair-repeat polymorphism and mood disorders: a meta-analysis. Biol Psychiatry. 2005 May 1;57(9):999-1003.
8. McGuffin P, Alsabban S, et.al.: The truth about genetic variation in the serotonin transporter gene and response to stress and medication. Br J Psychiatry. 2011 Jun;198(6):424-7.
9. Inoue K., Itoh K., et.al.: Positive association between T-182C polymorphism in the norepinephrine transporter gene and susceptibility to major depressive disorder in a japanese population. Neuropsychobiology. 2004;50(4):301-4.
10. S.P. Leighton, L. Nerurkar, et. Al.: Chemokines in depression in health and in inflammatory illness: a systematic review and meta-analysis. Molecular Psychiatry (2018) 23, 48–58.
11. Robert Dantzer, Jason C. O’Connor: From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008 Jan; 9(1): 46–56.
12. Piotr Gałecki, Monika Talarowska: Teoria zapalna depresji – najważniejsze fakty. Psychiatr. Pol. 2018; 52(3): 437–447.
13. Heather M. Derry, Avelina C. Padin: Sex Differences in Depression: Does Inflammation Play a Role?. Curr Psychiatry Rep. 2015 October ; 17(10): 78.
14. Lotte Gerritsen, Yuri Milaneschi, et.al.: HPA Axis Genes, and Their Interaction with Childhood Maltreatment, are Related to Cortisol Levels and Stress-Related Phenotypes. Neuropsychopharmacology (2017) 42, 2446–2455.
15. Abhay Sharma: Systems Genomics Support for Immune and Inflammation Hypothesis of Depression. Current Neuropharmacology, 2016, 14, 749-758.
16. Carmine M.Pariante: Why are depressed patients inflamed? A reflection on 20 years of research on depression,glucocorticoid resistance and inflammation. European Neuropsychopharmacology (2017) 27, 554–559.